
Next-generation proteomics technologies – Next-generation protein sequencing

Jamie Rose Kuhar
November 17, 2022
As we discussed in a previous post, proteins are the key determinants of cellular function. Recognizing this, scientists have been using and developing means to study proteins for many years. However, traditional methods of studying proteins cannot provide truly comprehensive data on the proteome, the full set of proteins in a cell with their associated abundances and locations. Even a relatively omics-scale method like mass spectrometry can only measure a small fraction of the proteins in a cell. Accessing largely untapped, comprehensive proteomic data will enable scientists to better understand and manipulate cellular functions in health and disease. This will lead to better disease treatments and diagnostics in addition to high-yielding crops, and novel means of protecting the environment.
Seeing this potential, many scientists have set out to create next-generation proteomics technologies. These technologies aim to deliver comprehensive data on the proteome from any sample of interest. Although many of these technologies are still in development, they should be available to researchers soon and will spur a proteomics revolution. In this pair of posts, we cover the concepts behind two kinds of next-generation proteomics technologies including:
- Next-generation protein sequencing
- Protein Identification by Short Epitope Mapping (PrISM)
These posts are not intended to provide in-depth descriptions, but instead provide an overview of how these technologies work.
In the graphics portraying the technologies below, we provide qualitative assessments of proteome coverage and ease-of-use. While technologies with low proteome coverage are generally used for targeted experiments looking at a small number of proteins at once, medium coverage technologies can look at 100s to 1000s of proteins at once. Truly comprehensive, high proteome coverage technologies can analyze tens of thousands of proteins at once.
Low ease-of-use technologies generally require complicated, difficult, or customized sample preparation with a lot of hands-on experimenter time, and their data may be difficult to analyze or require bioinformatics support. High ease-of-use technologies employ simple, standardized sample preparation, include more automation, and provide simple, data-rich outputs. Medium ease-of-use technologies have some mix of these attributes.
In addition to assessing standard metrics like these, we also discuss some of the pros and cons of each technology. Many of the pros and cons discussed below are specific to the capabilities of the technology.
Importantly, the technology assessments here should not be viewed as definitive. Rather, they aim to help you think about how you can best leverage these technologies for your experimental goals.
For more in-depth information on next-generation proteomics technologies see Alfaro et al 2021 and Timp and Timp 2020.
Check out this video to learn how we quantify proteins on the Nautilus Platform
Edman degradation-based next-generation protein sequencing
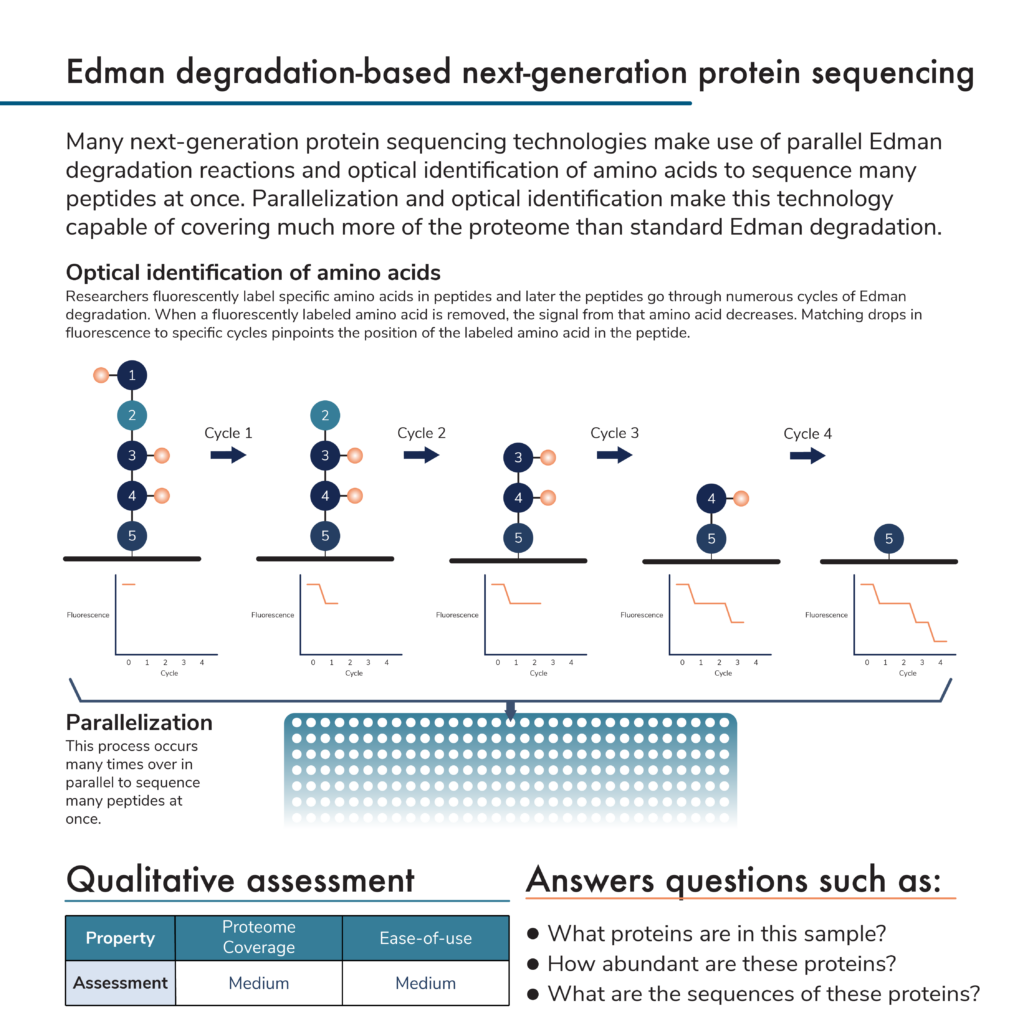
In a previous post, we covered the traditional protein sequencing technique known as Edman degradation. Many new protein sequencing methodologies build off of this technique. In brief, Edman degradation involves a series of chemical reactions that remove amino acids from one end of a protein or peptide sequentially. As the amino acids are removed, they are isolated and their particular identities can be determined using a variety of biochemical techniques. Scientists build peptide sequences from these amino acid identifications, and multiple peptide sequences are combined into full protein sequences.
Edman degradation itself is slow, but next–generation protein sequencing techniques automate the process to make it possible to sequence many peptides from multiple proteins at once. They perform many Edman degradation reactions in parallel and use optical methods to identify amino acids.
Optical identification of amino acids helps speed up the sequencing process. This can work in a few ways, but we focus on one here. Amino acids are fluorescently labeled in a peptide before the Edman degradation process begins. Optical equipment in protein sequencing instruments can measure the fluorescence intensity of various labels. The more labels there are for a particular amino acid, the more intense the signal. As the Edman degradation process proceeds, individual amino acids will be removed from the peptide and, if they are labeled, the fluorescence intensity from that label will decrease. By counting the number of degradation cycles it takes to decrease the fluorescence of a particular kind of labeled amino acid, the instrument can pinpoint the amino acid’s location in the peptide.
Currently, researchers cannot make fluorescent compounds that specifically label every amino acid in a peptide, which limits coverage, and the approach is peptide based, which can limit dynamic range. Nonetheless, with information for a subset of amino acids and databases of known protein sequences, these techniques can identify substantially more proteins at once than traditional Edman degradation.
Pros and cons of Edman degradation-based next-generation protein sequencing
Pros
- Novel protein identification – Techniques like these could theoretically sequence proteins without needing to reference any databases or at least without referencing pre-established outputs from the instrument.
- Single–molecule measurements – These technologies will provide data at the single–peptide level as opposed to the aggregated data provided by many techniques. Therefore, they should be more sensitive. With enough reactions performed in parallel, next-generation sequencing technologies should provide researchers with a more comprehensive look at the proteome.
Cons
- Throughput – Each cycle of Edman degradation is time consuming and sequencing full peptides, even in parallel, may take longer than a day.
- Limited ability to decipher protein modifications – Individual amino acids in proteins are modified in many ways including the addition of sugar molecules, phosphate groups, and other chemicals. The result is a wide variety of proteoforms that can have drastically altered functions. Unfortunately, researchers cannot label all modified amino acids and some modifications prevent the Edman degradation process from proceeding. Thus, next-generation sequencing is blind to many modifications, and modifications can decrease the amount of information that researchers get from the process.
- Peptide length – This technique sequences short peptides and not full proteins. Peptide-based sequencing may limit dynamic range as multiple peptides will contribute to a single protein and need to be sequenced independently.
- Peptide-based inference of protein identity – This technique infers protein identity by compiling the sequences of multiple component peptides. Such inferences can be inaccurate if multiple proteins species in a sample are composed of similar peptides.
Next-generation sequencing has the potential to build protein sequences directly from individual amino acids but may not always provide comprehensive protein identifications or a full picture of protein modifications. Below we describe how protein sequencing using pore-based methods can provide more information. While pore-based sequencing technologies are promising, they are in the early stages of development and so we do not provide a full assessment of their pros and cons below.
Pore-based protein sequencing
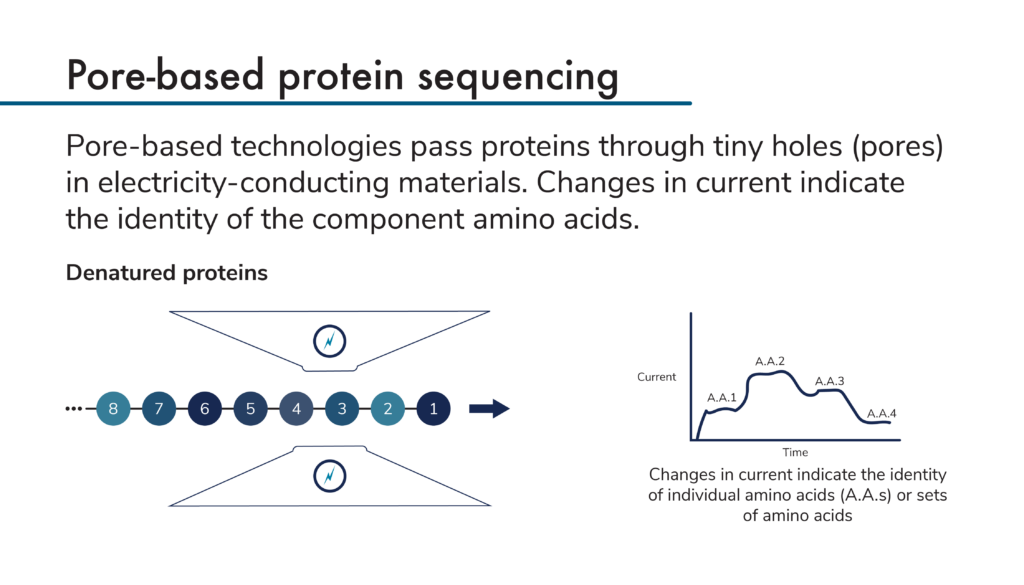
Pore-based protein sequencing technologies all make use of electricity-conducting materials that have tiny pores through which proteins can pass. As proteins pass through the pores, they cause changes in the current flowing through the materials. Measurements of the changing current create signatures that can be used to identify the proteins passing through the pores.
To use these technologies to sequence proteins, researchers first denature and linearize their proteins. Then, they pass the proteins through pores, either amino acid by amino acid or a few amino acids at a time. As the amino acids in the protein pass through the pore, they cause characteristic changes in current that depend on the identities of each individual amino acid. Modified amino acids produce their own characteristic changes in current as well. Theoretically, it should be possible to directly sequence proteins in this way.
As with Edman degradation-based protein sequencing, pore-based technologies must be parallelized to achieve proteome-level coverage in a reasonable amount of time. Engineering techniques and nanofabrication methodologies are being leveraged to create arrays of the small pores required by these techniques. Using these arrays, scientists should be able to identify many proteins in a high-throughput way.
Pore-based technologies are incredibly promising but require more development and so we do not provide a standardized assessment of them here. In our next post, we will cover Protein Identification by Short epitope Mapping (PrISM). This is the theoretical framework behind the Nautilus platform and will provide simple but information-rich outputs that can be applied to a wide range of research projects in the near-term.
Explore the future of proteomics
Check out this episode of our Translating Proteomics podcast to learn about recent developments in proteomics
MORE ARTICLES
