

The proteome is the full set of proteins within a biological sample like a cell, tissue, or organism. Proteomics studies all of these proteins and their interactions through various protein analysis methods.
The particular proteomic make-up of any biological entity, including individual protein abundances and locations, determines how that entity works at the molecular level. Thus, mapping a cell’s proteome provides mechanistic information on biological activities and makes it possible to study the mechanisms behind these activities as well as disease states in human health. Knowledge of the proteome may lead to a broad range of discoveries and applications including everything from new therapeutics to new ways to protect plants from drought.
Given the essential importance of the proteome, scientists have been studying it for many years using various protein analysis methods like mass spectrometry, protein sequencing and more. In this “Traditional protein analysis” series, we cover how researchers typically study the proteins that make up the proteome.
These protein analysis methods are not necessarily “ omics” scale technologies, which generally aim to measure all or a large amount of a particular type of biological molecule in a sample. Nonetheless, these methods form the foundations upon which proteomic analysis methods have been built. In a future series, we will dive into the technical concepts behind emerging omics technologies, which aim to enable scientists to study the full proteome.
Watch out our animation to learn how we quantify proteins on the NautilusTM Proteome Analysis Platform
Comparing different protein analysis methods
In the graphics portraying the technologies below, we provide qualitative assessments of proteome coverage and ease-of-use for each. Technologies with low proteome coverage, like western blotting and flow cytometry, are generally used for targeted experiments analyzing a small number of proteins at once. Medium coverage technologies like affinity arrays can look at 100’s to 1000’s of proteins in a single experiment. Truly comprehensive, high proteome coverage technologies can analyze tens of thousands of proteins at once.
Some protein analysis methods are easier to use than others. Low ease-of-use protein analysis technologies generally require complicated, difficult, or customized sample preparation involving a lot of hands-on experimenter time, and their data may be difficult to analyze or require bioinformatics support. High ease-of-use protein analysis technologies employ simple, standardized sample preparation, include more automation, and provide simple, data-rich outputs including protein abundance. Medium ease-of-use technologies have some mix of these attributes.
In addition to assessing standard metrics like these, we also discuss some of the advantages and disadvantages of each protein analysis technique. Many protein profiling technologies have specific pros and cons that make them ideal for certain applications, but not others.
Importantly, the technology assessments here should not be viewed as definitive. Rather, our goal is to help you think about how you can best leverage these technologies for your specific experimental goals.
The fundamental work of protein sequencing
Delving into the proteome starts with exploring proteins themselves. For instance, a scientist may find cells that carry out a particular function and want to know what protein or proteins give them that function. To figure this out, they can extract the proteins from the cells and separate the proteins in fractions based on a variety of characteristics such as size and hydrophobicity. Then, a researcher can test each protein fraction to see if it still has the function of interest. If it does, the researcher can repeat the process, making the protein fractions smaller and smaller, hopefully isolating an individual protein with the function they’re interested in.
Even after isolating a protein, researchers still don’t know the identity of the protein, what gene encodes it, or how that protein carries out its function. That’s where protein sequencing methods come in.
Proteins are composed of chemical building blocks called amino acids. There are 20 amino acids with a variety of chemical properties. These are attached to one another in a linear fashion to form full proteins, and their precise order and abundance in a protein gives the protein its specific structure and function. Protein sequencing methods can determine the order and abundance of all the amino acids that make up a protein.
Sequencing proteins can give scientists information about:
- Protein identity – A protein’s sequence determines its identity. The sequence of amino acids in a protein ultimately determines how it folds in 3D space and gives the protein its particular shape, and function. Once researchers sequence a protein, it can be identified.
- What gene encodes the protein – Individual amino acids are encoded by codons, three letter combinations of the four nucleic acids in DNA (A, G, C, and T). If researchers know the amino acid sequence of a protein, they can search through the genome of the organism the protein came from to find any DNA sequences (genes) capable of encoding that protein. Knowing the gene and its location in the genome may provide clues about the production of the protein. Knowing the gene that encodes a protein also enables researchers to direct cells to produce that protein. Rather than go through an arduous purification process every time they want to study a protein, with a gene in hand, researchers can instead instruct cells to produce the protein in large amounts or a in a way that makes that protein much easier to purify.
- Protein function – Knowing a protein’s sequence gives researchers more information about its function. In addition, computational methods for determining a protein’s structure based on its amino acid sequence have improved phenomenally over the last couple of decades (Jumper et al 2021). Using modern methods, scientists can compare amino acid sequences and structures across proteins to make hypotheses about protein function with confidence.
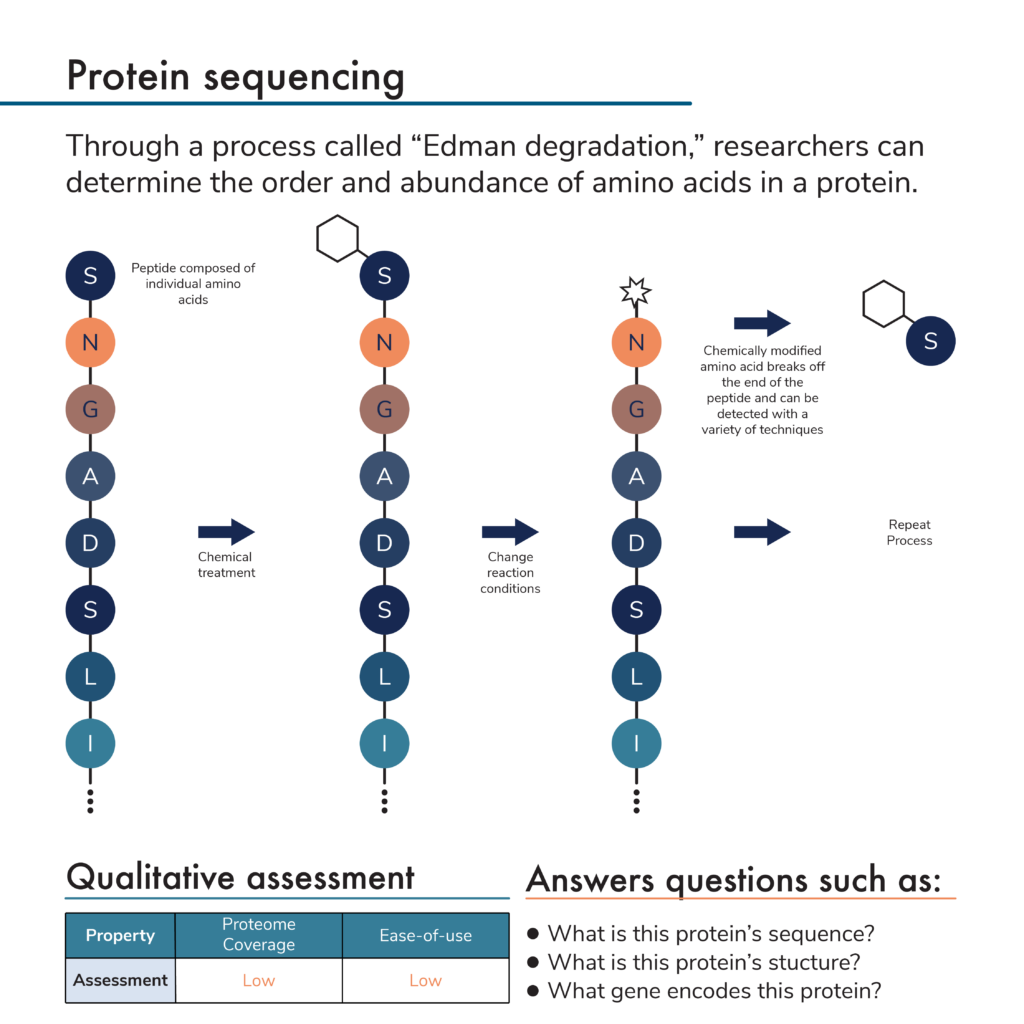
Protein sequencing by Edman degradation
So how do researchers determine protein sequences? Traditionally, protein sequencing is done with Edman degradation. In this protein sequencing method, a purified protein is incubated with a chemical that attaches to an amino acid on one of its ends. Manipulating reaction conditions causes this chemical and the terminal amino acid to break off. The chemical-terminal amino acid compound can then be extracted from the rest of the protein.
Researchers can use a variety of analytical techniques to determine which of the 20 amino acids is contained in the extracted compound. Repeating this process many times over reveals the full amino acid sequence of a protein and thus its identity.
The Edman degradation protein sequencing method is very slow and can only be used to sequence short fragments of full proteins called peptides. Nonetheless, by breaking apart a purified protein and sequencing its component peptides, researchers can determine the full sequence of the original protein.
Advantages of Edman degradation
- Novel protein sequencing – Using Edman degradation, researchers can sequence newly purified proteins without any prior information about them.
Disadvantages of Edman degradation
- Proteome coverage – Before sequencing proteins using Edman degradation, researchers must first isolate them. This makes it difficult to sequence many proteins at once. In addition, it’s only possible to sequence one peptide at a time in a single reaction. Finally, certain post-translational modifications of the peptide sequence can interfere with the chemical reactions involved in Edman degradation, limiting its use on some fraction of the proteome.
- Peptide-based inference of protein identity – This technique infers a protein sequence by compiling the sequences of multiple component peptides. Such inferences can be inaccurate if multiple protein species in a sample are composed of similar peptides.
- Low throughput – The Edman degradation protein sequencing process is slow. Automation can speed up the process, but it will still take about a day to get a full protein sequence.
- Low ease-of-use – Preparing proteins for Edman degradation can be time consuming and difficult. While the Edman degradation process can be automated, data analysis can be difficult because many peptides have similar sequences and it may not be clear how they should be combined into a full protein sequence.
Researchers are currently developing variations on Edman degradation to sequence many proteins at once. While Edman degradation is slow, one of its key advantages is it does not require prior knowledge of a protein’s sequence to determine its full identity.
Antibodies, on the other hand, require known, well-validated protein targets to be useful for protein identification. And, as you’ll learn in the next post in this series, mass spectrometry relies on prior knowledge of proteins to determine protein sequence as well. Thus, while protein sequencing may be a bit old school, it’s still very useful when researchers have limited information!
MORE ARTICLES
