
Traditional protein analysis methods – Mass spectrometry proteomics

Tyler Ford
November 29, 2022
Mapping a cell’s proteome, the collection of proteins and proteoforms it contains, is a vital part of proteomics. The work of proteomic analysis can provide detailed information on biological activities and make it possible to study the mechanisms behind these activities as well as diseases associated with them. One of the most common ways to do this today is mass spectrometry proteomics, which includes a number of different techniques that researchers can use to explore the proteome.
The proteomic knowledge gained from these techniques may lead to a broad range of discoveries and applications including everything from new therapeutics to new ways to protect plants from drought.
Given the essential importance of the proteome, scientists have been using various protein analysis methods like Western blotting, protein sequencing and more to conduct proteomic analyses for years. In this “Traditional protein analysis” series, we cover how researchers typically study the proteins that make up the proteome, including through mass spectrometry.
These protein analysis methods are not necessarily “omics” scale technologies, which generally aim to measure all or a large amount of a particular type of biological molecule in a sample. Nonetheless, these methods form the foundations upon which proteomic analysis methods have been built. In a separate series on next-generation proteomics technologies, we dive into the technical concepts behind emerging proteomics technologies, which aim to enable scientists to conduct proteomic analyses of the full proteome.
How does a mass spectrometer work for proteomics?
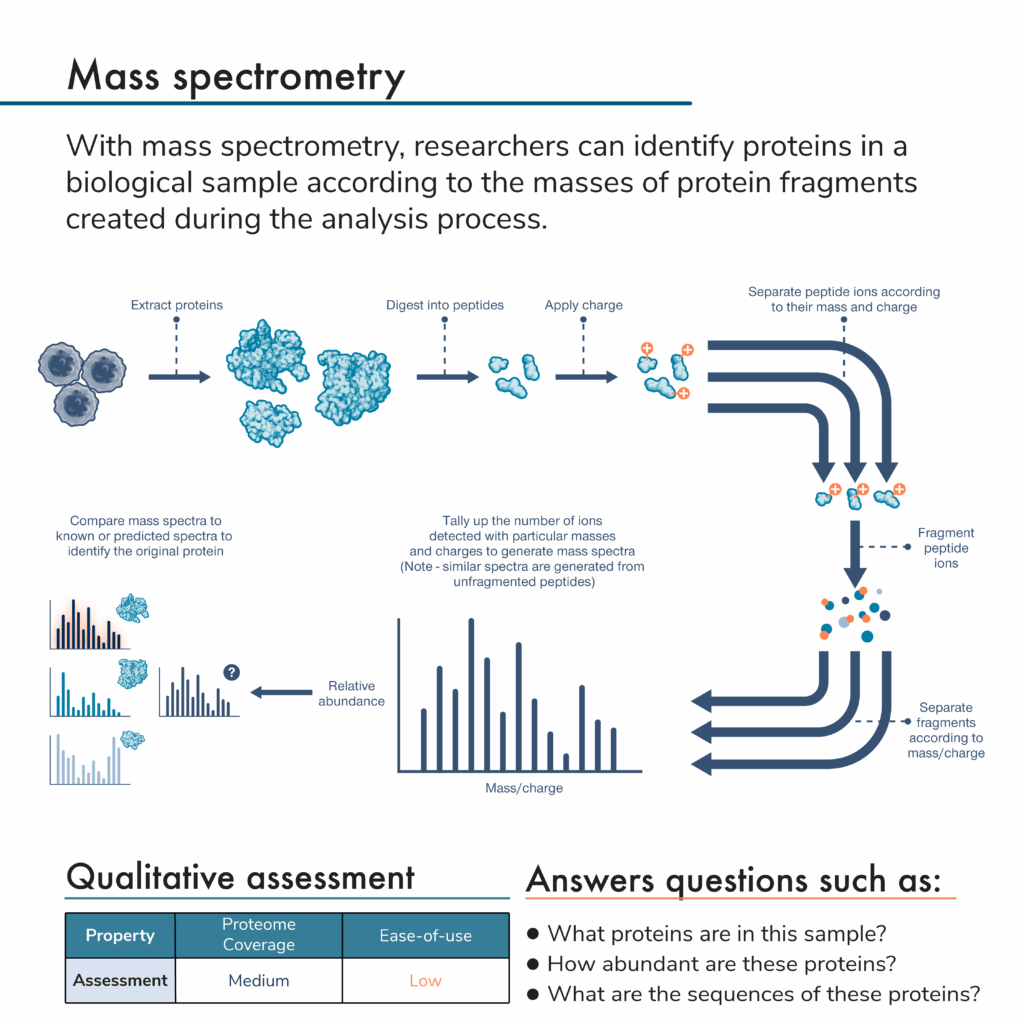
While there are many versions of mass spectrometry that can be used for proteomic analysis, they generally involve:
- Extracting proteins from a biological sample
- Breaking the proteins into peptides
- Giving the peptides an electric charge
- Passing the charged peptides through a device inside a mass spectrometer where they travel at a certain rate based on their mass and charge
- Breaking separated peptides apart into small fragments
- Passing the fragments through a device that separates them according to mass and charge again
- Allowing the separated fragments to be detected and generating spectra, which reveal the number of fragments of different masses and charges that are detected
- Computational matching of the generated spectra to known or predicted spectra for specific proteins allowing for protein identification
Applications of mass spectrometry for proteomic analysis
Mass spectrometry can be used for multiple kinds of proteomic analysis, including discovery proteomics, targeted proteomics, and more. Proteomics researchers can also use mass spectra to measure the abundance of the identified proteins.
As you might imagine, some proteins can generate very similar spectra and/or may not be separated well because they have similar masses and charges. In addition, protein spectra signals from high abundance proteins can often drown out signals from low abundance proteins. Thus, even with extensive optimization, mass spectrometry can only measure a fraction of the proteome.
Researchers can complement mass spectrometry with a variety of techniques that make the protein spectra easier to analyze. For instance, before putting them through the mass spectrometer, researchers can pass their proteins or digested proteins (peptides) through separation columns. These columns are tubes containing porous materials that separate proteins or peptides based on properties like size or hydrophobicity. Separations can be accomplished off-line, where a single sample is separated into multiple fractions and each fraction is analyzed separately. Alternatively, with in-line separation, a researcher adds a single sample to the separation column and the proteins or peptides exiting the column can directly enter a mass spectrometer. Protein or peptide analytes are sequentially analyzed by the mass spectrometer based on how they interact with the column. As only a fraction of the proteins or peptides in the sample exit the column at any point in time, the mass spectrometer can focus analysis on a smaller number of analytes increasing detection and quantitation resolution.
While there are many mass spectrometry techniques for proteomics that can ultimately make the resulting spectra easier to analyze, they introduce biases into the protein detection process. For instance, some proteins may be incompatible with certain columns. Thus, these time-consuming processes may limit the amount of data researchers get from proteomic analysis using mass spectrometry in one experiment.
Advantages of mass spectrometry for proteomic analysis
- Omics-scale – While mass spectrometry cannot provide measurements of the full proteome, it can provide data on thousands of proteins at once, making it useful for broadscale proteomics. This technique provides more comprehensive information than the other techniques discussed in this series and makes mass spectrometry a better option for proteomics research than other traditional methods.
- Can detect peptide modifications – Although the data analysis can be difficult, mass spectrometry can be used to identify proteins that have been modified through biological processes such as phosphorylation and glycosylation. These modified proteins are known as proteoforms.
Disadvantages of mass spectrometry for proteomic analysis
- Low dynamic range – Signals from high abundance proteins can drown out those from low abundance proteins, making potentially very important proteins difficult to analyze.
- Peptide-based inference of protein identity – With mass spectrometry, protein identification involves compiling sequences of multiple component peptides. Inferring full protein sequences from peptides can be inaccurate if multiple protein species in a sample are composed of similar peptides. Peptide-based inference also makes it impossible to identify proteoforms – the single-molecule variants of proteins with their specific sets of modifications found in biological systems. There are efforts to improve “top-down” proteomics methods that analyze intact proteins and retain such information, but these are not widely used today.
- Cost – Mass spectrometers are very expensive, making them less ideal for smaller laboratories doing protein analysis.
- Low ease-of-use – Protein preparation for mass spectrometry may need to be highly customized depending upon the sample type and what fraction of the proteome researchers wish to study. Mass spectrometers themselves also have a high learning curve and the spectra they generate must be analyzed by specialized software.
The role of mass spectrometry in proteomics research
Despite its limitations for proteomics, mass spectrometry has been, for many years, the main tool available for protein identification, broadscale proteomics, targeted proteomics, and more. The technique has been used to make many biological discoveries and develop many therapeutics. Mass spectrometry is continually being improved through instrumentation developments, innovative methods, and improved software for data analysis.
To overcome some of the drawbacks of mass spectrometry for proteomics, many researchers are developing new technologies that will make it easier to comprehensively analyze the proteome. We discuss the concepts behind some of these up-and-coming proteomics technologies, including our own platform, in other “Proteomics” posts.
Learn about other traditional protein analysis methods like protein sequencing and antibodies and other affinity reagents in the previous two posts in this series.
Explore the future of proteomics
Watch the video below to learn how the Nautilus Proteome Analysis Platform is designed to comprehensively quantify the proteome
Notes on comparing different protein analysis methods
In the graphics portraying the technologies throughout this series, we provide qualitative assessments of proteome coverage and ease-of-use for each. Technologies with low proteome coverage, like western blotting and flow cytometry, are generally used for targeted experiments analyzing a small number of proteins at once. Medium coverage technologies like affinity arrays can look at 100’s to 1000’s of proteins in a single experiment. Truly comprehensive, high proteome coverage technologies can analyze tens of thousands of proteins at once.
Some protein analysis methods are easier to use than others. Low ease-of-use protein analysis technologies generally require complicated, difficult, or customized sample preparation involving a lot of hands-on experimenter time, and their data may be difficult to analyze or require bioinformatics support. High ease-of-use protein analysis technologies employ simple, standardized sample preparation, include more automation, and provide simple, data-rich outputs including protein abundance. Medium ease-of-use technologies have some mix of these attributes.
In addition to assessing standard metrics like these, we also discuss some of the advantages and disadvantages of each protein analysis technique. Many protein profiling technologies have specific pros and cons that make them ideal for certain applications, but not others.
Importantly, the technology assessments here should not be viewed as definitive. Rather, our goal is to help you think about how you can best leverage these technologies for your specific experimental goals.
MORE ARTICLES
