Applications of proteomics in immunology – Outcomes of ubiquitylation in activated CD4+ T cells

Tyler Ford
January 4, 2024
Many proteins are altered via post- translational modifications. These are wide ranging and can include the addition of modifying groups such as phosphates, sugars, and small proteins as well as protein cleavage. Such modifications, which create variants of proteins known as proteoforms, have many effects on protein and cellular function. The human proteome contains millions of proteoforms, greatly expanding the number of potential protein functions in the body. One powerful application of proteomics research is finding and studying proteoforms, especially those known to be involved in disease.
For example, ubiquitylation is a post translational modification involving the addition of a protein called ubiquitin to target proteins. This modification is important for many processes including regulating immune responses. Ubiquitylation is well studied for its effects on protein degradation and there have been several studies on how ubiquitylation regulates T cell activation via protein degradation.
However, ubiquitylation also has a slew of lesser-studied non-degradative effects on proteins. These include changing protein function, protein localization, and protein-protein interactions. Recently, a study leveraging proteomics and other approaches by Dybas et al. found an increase in non-degradative ubiquitylation in activated CD4+ T cells, which release cytokines to mediate the immune response. Once activated, the CD4+ T cell proteome undergoes significant changes in protein expression.
Below we discuss how these researchers leveraged a multiomics approach to study ubiquitylation and how innovative proteomics technologies, such as the NautilusTM Proteome Analysis Platform, can advance studies involving complex proteoforms like these.
Studying ubiquitylation with transcriptomics, whole cell proteomics, and di-glycine remnant proteomics
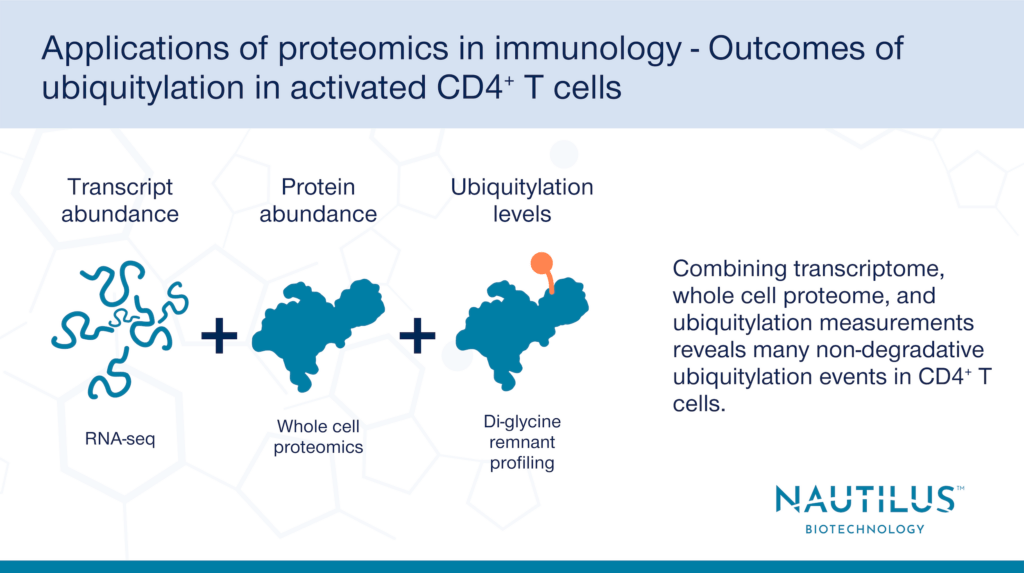
To measure the effects of proteome-wide ubiquitylation, Dybas et al. used a combination of di-glycine remnant proteomics, whole cell proteomics, and transcriptomics in “rested” vs. “activated” CD4+ T cells. Why the multiomic approach? Just one method alone does not give the entire picture of protein ubiquitylation and its outcomes.
Di-glycine remnant profiling, where proteins with ubiquitylated lysine residues are digested into fragments and identified by mass spectrometry, by itself identifies ubiquitylation sites, but does not determine the effects of ubiquitylation on protein abundance. Whole cell proteomics reveals changes in protein abundance but does not show whether changes in abundance are due to post translational modifications like ubiquitylation or due to changes in transcript abundance. Lastly, transcriptomics alone reveals changes in transcript abundance, but these changes don’t necessarily correlate with similar changes in protein abundance or post-translational modifications.
By leveraging a multiomics approach and using all three techniques in tandem, Dybas et al. were able to determine whether ubiquitylation correlates with a degradative or non-degradative outcome for ubiquitylated proteins after CD4+T cell activation.
For example, to determine whether protein abundance changes are a result of changes to transcription as opposed to post translational modification, researchers can use transcriptomics to determine how transcript changes correlate with protein abundance differences. If transcripts increase or decrease along with protein abundance, it’s evidence that protein levels are being determined by transcript levels. However, if changes in protein abundance are not correlated with changes in transcript levels, post translational modifications may be impacting protein levels. In fact, after conduction protein quantification, Dybas et al. found that one third of the proteins in their studies have abundance levels that changed in the opposite direction of their transcripts. This indicates that post translational modifications may drive the observed proteomic difference.
Examining whole cell proteomics data alongside di-glycine remnant profiles can help researchers determine whether ubiquitylation has a degradative effect on a protein. For example, cases where a protein’s abundance decreases based on whole cell proteomics but its di-glycine remnants’ abundance increase likely indicate degradative ubiquitylation. If both protein abundance and ubiquitylation change in the same direction (ex: both increase), it shows that the change in ubiquitylation levels may be due to changes in protein levels caused by other factors such as changes to transcription and not degradative ubiquitylation.
Degradative and non-degradative ubiquitylation after T cell activation
Dybas et al. focused their multiomic analysis on a set of 155 T cell receptor (TCR) signaling proteins as ubiquitylation has known roles in TCR signaling. 12 of these proteins had a greater than 25% increase in ubiquitylation in activated cells vs. rested cells. Four of these proteins had ubiquitylation patterns indicative of degradative ubiquitylation (ex: increased ubiquitylation but decreased protein abundance). The other eight proteins had similar changes in transcriptome and protein abundance, suggesting gene expression was driving the change in protein abundance and ubiquitylation was non-degradative.
To investigate how different types of ubiquitylation affect proteins, the researchers used targeted proteomics to examine how ubiquitin was attached to the proteins that had degradative and non-degradative outcomes. Ubiquitin has seven lysine residues that it can use to attach to a target protein. The researchers found an increased use of the K29, K33, and K63 residues of ubiquitin in activated CD4+ T cells compared to rested CD4+ T cells. As K29, K33, and K63 ubiquitylation are known to be associated with non-degradative ubiquitylation, it appears that CD4+ T cell activation is associated with an increase in non-degradative ubiquitylation. Remnant peptides associated with proteasomal degradation (K48 and K11 ubiquitylation), on the other hand, were unchanged in rested vs. activated CD4+ T cells.
In total, the researchers multiomic analysis found over 1,200 proteins were ubiquitylated in CD4+ T cells out of over 5,500 proteins identified using whole cell proteomics. As ubiquitylated proteins thus accounted for over 20% of total proteins detected, there is a wealth of opportunity to further study the role of ubiquitylation in T cell activation. The dataset from Dybas et al. provides a good starting point for further investigation.
Applications of next-generation proteomics in studying ubiquitylated proteoforms
Post-translational modifications such as ubiquitylation are an important part of cell signaling. Ubiquitylation manifests in a variety of modifications and this complexity makes it challenging to identify and quantify various ubiquitylated proteoforms. In the study discussed above, proteins had either single or multiple ubiquitylation sites and ubiquitin was attached via one of seven different possible lysine residues. This is a salient illustration of the complexity underlying ubiquitylation.
It can be difficult to analyze rare proteoforms among abundant proteins with traditional proteomics platforms. Because it is designed with a high dynamic range and ability to analyze billions of intact proteins at the single-molecule level, the Nautilus Proteome Analysis Platform could help immunologists and other scientists establish more in-depth insights into proteoforms present in their cells of interest. With our platform, we hope researchers will be able to link proteoforms to important functional outcomes in health and biology in general.
MORE ARTICLES
