
Nautilus™ Proteome Analysis Platform
Revealing the depths of the proteome with unprecedented scale and accessibility

Quantify the entire proteome

Building a platform to power next-generation proteomics
With current technologies, researchers can only measure 8-30% of the proteome routinely and must use complicated, customized workflows to do so. Frustrated with the status quo, we set out to build a next-generation proteomics platform that can quantify the whole proteome with an integrated workflow that is accessible to a wide range biological research labs.
Advantages of the Nautilus™ Proteome Analysis Platform

Comprehensive proteome
coverage
Unleash >95% of the proteome for holistic insights into the biology of your samples.
Learn more
Comprehensive proteome
coverage
Multi-affinity probes enables quantification of 1,000s of unique proteins providing high quality data throughput per sample.

Wide dynamic
range
Match the scale of the proteome with up to 10 billion single-molecule protein measurements per run.
Learn more
Wide dynamic
range
Up to 10 billion landing pads per sample provide up to 9 orders of magnitude dynamic range and limit the need for depleting high abundance proteins.

High
sensitivity
Identify and quantify proteins and proteoforms at the single-molecule level.
Learn more
High
sensitivity
Single-molecule counting provides the highest sensitivity for detection and quantification of proteins and proteoforms.

Reproducible and
robust
Quantitative, digital readout of intact, full-length proteins gives you confidence in your data.
Learn more
Reproducible and
robust
The combined use of hundreds of multi-affinity probes, a machine learning algorithm that parses binding data into high probability protein identifications, and automated workflows minimizes errors and improves reproducibility.

Rapid run
time
Speed time to discovery with an integrated workflow from sample to data.
Learn more
Rapid run
time
Delivering digital counts with a simple workflow avoids lengthy, time-consuming data analysis that requires specialized expertise.

Accessible
Any lab can measure the proteome with our platform.
Learn more
Accessible
Molecular labs can easily generate proteomic data from complex biological samples with simple workflows and automated data analysis and interpretation.
One platform, two analysis methods

Broadscale discovery proteomics
Our platform is designed to leverage a methodology known as Protein Identification by Short-epitope Mapping (PrISM) to analyze substantively the entire proteome of any sample using hundreds of proprietary multi-affinity probes. With this type of broadscale proteome analysis, researchers can perform discovery proteomics and learn how the proteome is affected in an unbiased way. In fact, modeling work shows that we can measure greater than 95% of the proteome in a variety of sample types using 300 cycles of multi-affinity probe binding. This should make it possible for scientists in any lab to routinely interrogate changes to the proteome under a variety of conditions in an unbiased manner.

Targeted proteoform analysis
Nautilus proteoform analysis will enable the quantification of protein isoforms and proteins with post-translational modifications. This is only possible on a platform capable of single-molecule measurements of intact proteins. This will complement top-down mass spectrometry approaches that are well-suited to de novo proteoform identification, but too complex and time consuming for exploring proteoform function across many samples.
Integrated workflow from sample to protein quantification
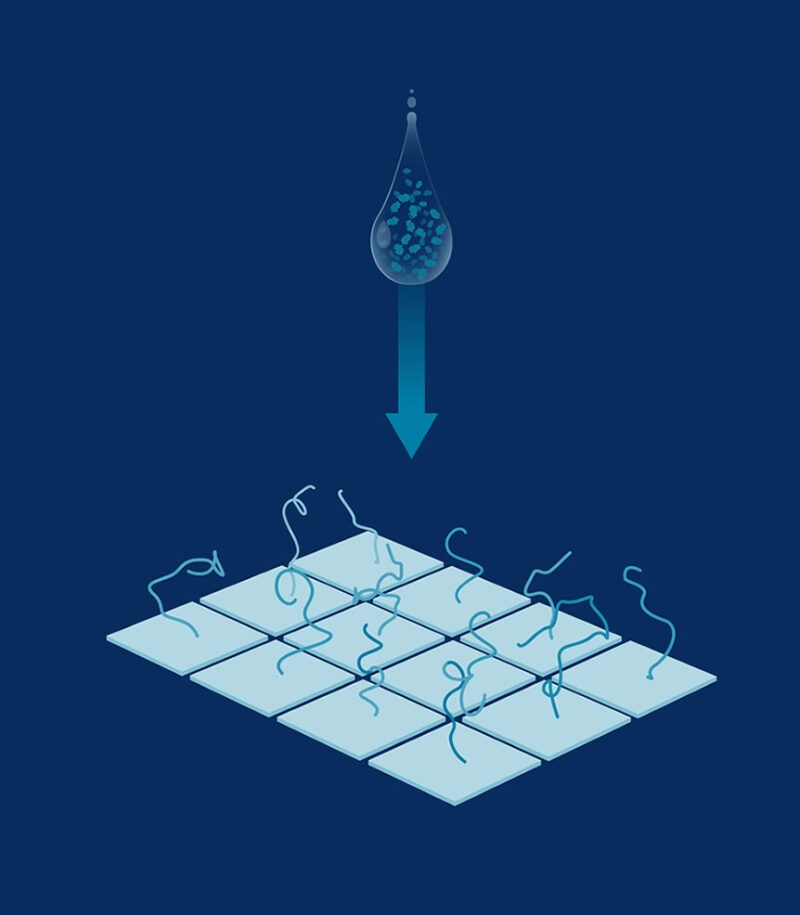
Protein library preparation
Proteins are functionalized to produce a library of single-molecules on a hyper-dense nanoarray.
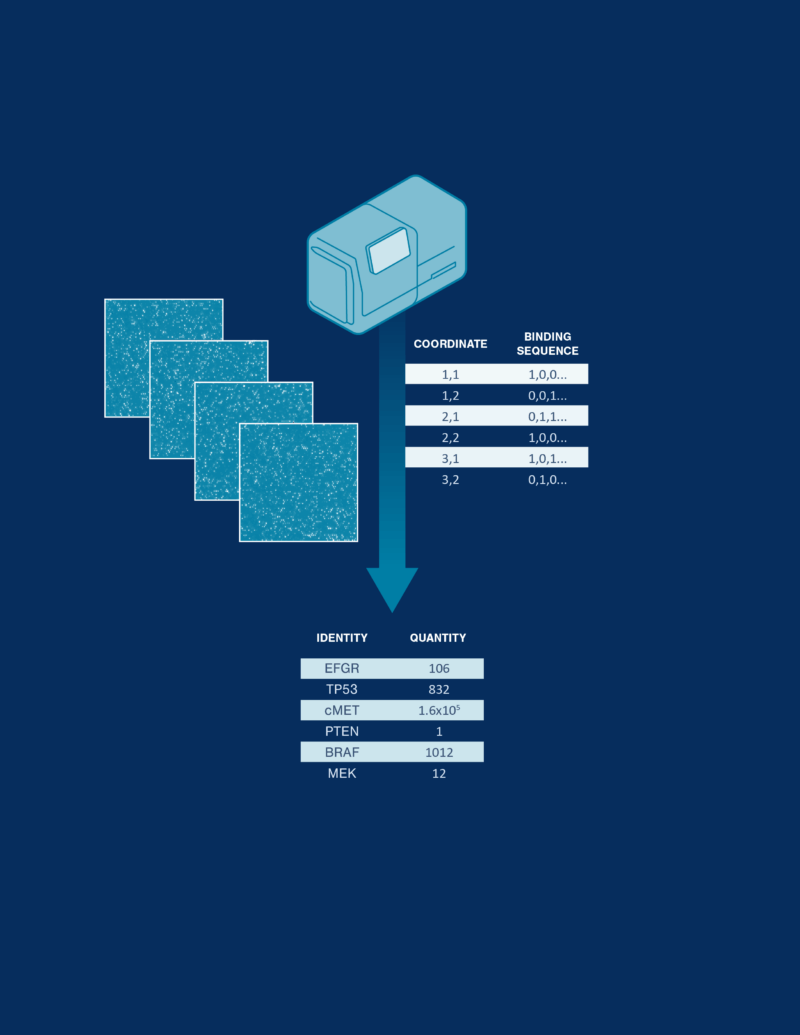
Decoding and quantification
Fluorescently labeled multi-affinity probes interrogate single proteins on the array and machine learning transforms each pattern of binding into protein identity and quantity.

Digital protein counts can be analyzed to discover new biology
The platform generates a simple output format with protein identities and counts. Data can be further analyzed in Nautilus’ cloud portal to derive biological insights.
Protein library preparation
Proteins are functionalized to produce a library of single-molecules on a hyper-dense nanoarray.
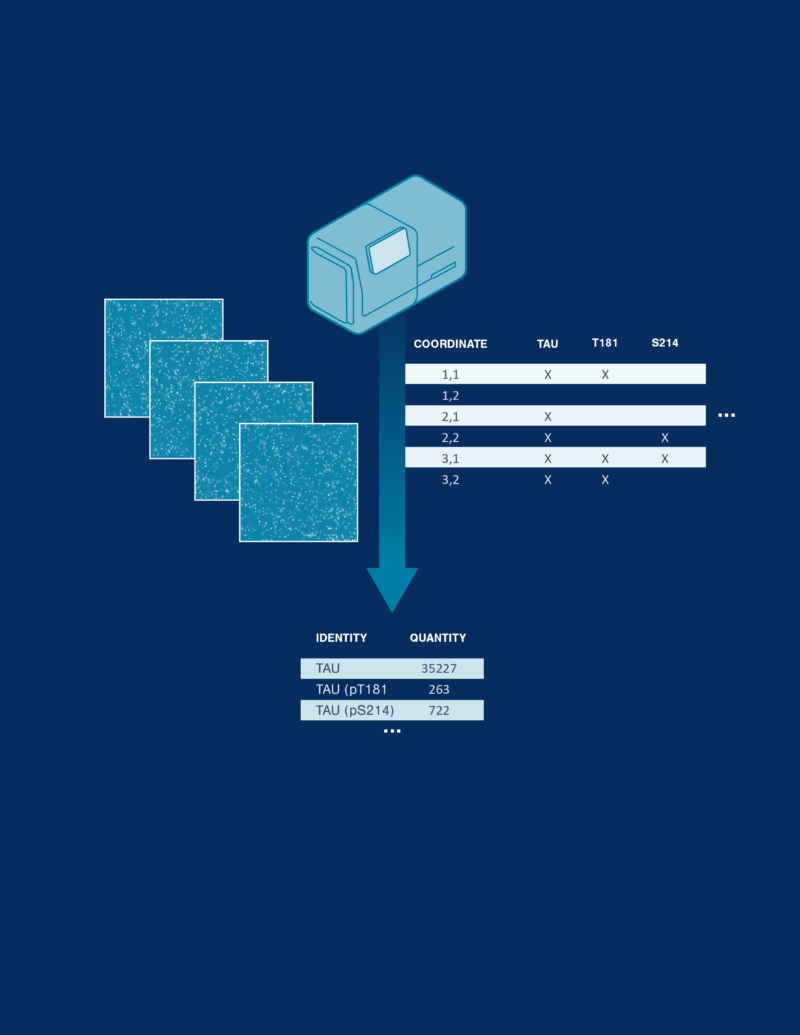
Proteoform identification and quantification
Targeted probes specific to proteoforms of interest are used to identify proteoforms on the platform. Quantification is performed by counting how many molecules of each proteoform are observed.
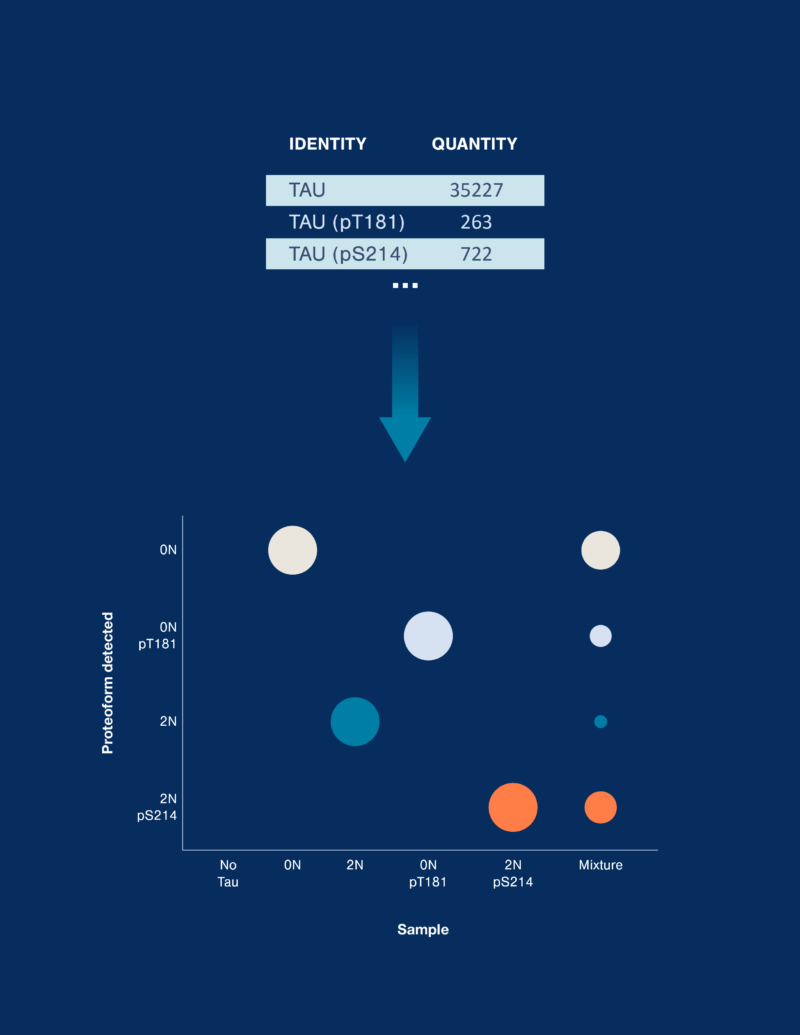
Data interpretation
Quantified proteoforms are analyzed with a variety of visualization tools to reveal new insights.
Driving biological discovery

Are you looking to unleash the power of the proteome?
Dive deeper into the science behind our platform and its applications







