


Proteomics has the incredible potential to reveal the mechanisms underlying biology. In doing so, it may provide the raw materials needed to develop the next generation of more effective biomarkers, diagnostics, and drug targets. In short, proteomics can revolutionize biomedicine.
However, without the help of AI, achieving these aims will be slow-going. In this blog series, we discuss:
- Why proteomics needs AI, and why we need better proteomic data to train models of biology
- How Nautilus data is designed to be well-suited for AI integration
- How Nautilus’ AI-ready data may improve multiomic analyses
Today’s post covers why proteomics needs AI and why we need better proteomic data to train models of biology.
Learn more about AI and biotech on the Translating Proteomics podcast.
Why does proteomics need AI?
Proteins are complicated. They come in many different shapes and sizes and have wildly diverse chemical properties – some are water soluble, some not, some are structurally inflexible, others not, some have many highly reactive modification sites, others do not. This diversity gives proteins the varied functions that make life work, but also makes them difficult to understand. There are simply too many nuances in any given proteome for a human to easily glean how proteins give cells their functions. On top of that, proteomes are dynamic and respond to changes in cellular conditions that fluctuate from second to second.
This all means that to understand proteomes, we need technologies that can resolve their nuanced differences over time. Given the large number of proteins in the proteome, their many possible proteoforms, and their ever-changing abundances, in most cases it will be incredibly difficult for humans to perceive and track patterns in proteomic fluctuations.
For a concrete example of this complexity, consider the heatmap of tau proteoforms shown below. This heatmap comes from our recent preprint. It shows how the levels of many tau proteoforms compare across samples from people with and without Alzheimer’s disease. While hierarchical clustering of this data was enough to deliver the exciting preliminary result that tau proteoforms profiles may differentiate individuals with and without Alzheimer’s and may correlate with disease severity, remember that these are profiles of just one protein’s proteoforms taken at a single time point. These results are likely as clear as they are because decades of research have already associated tau and its modifications with Alzheimer’s disease.
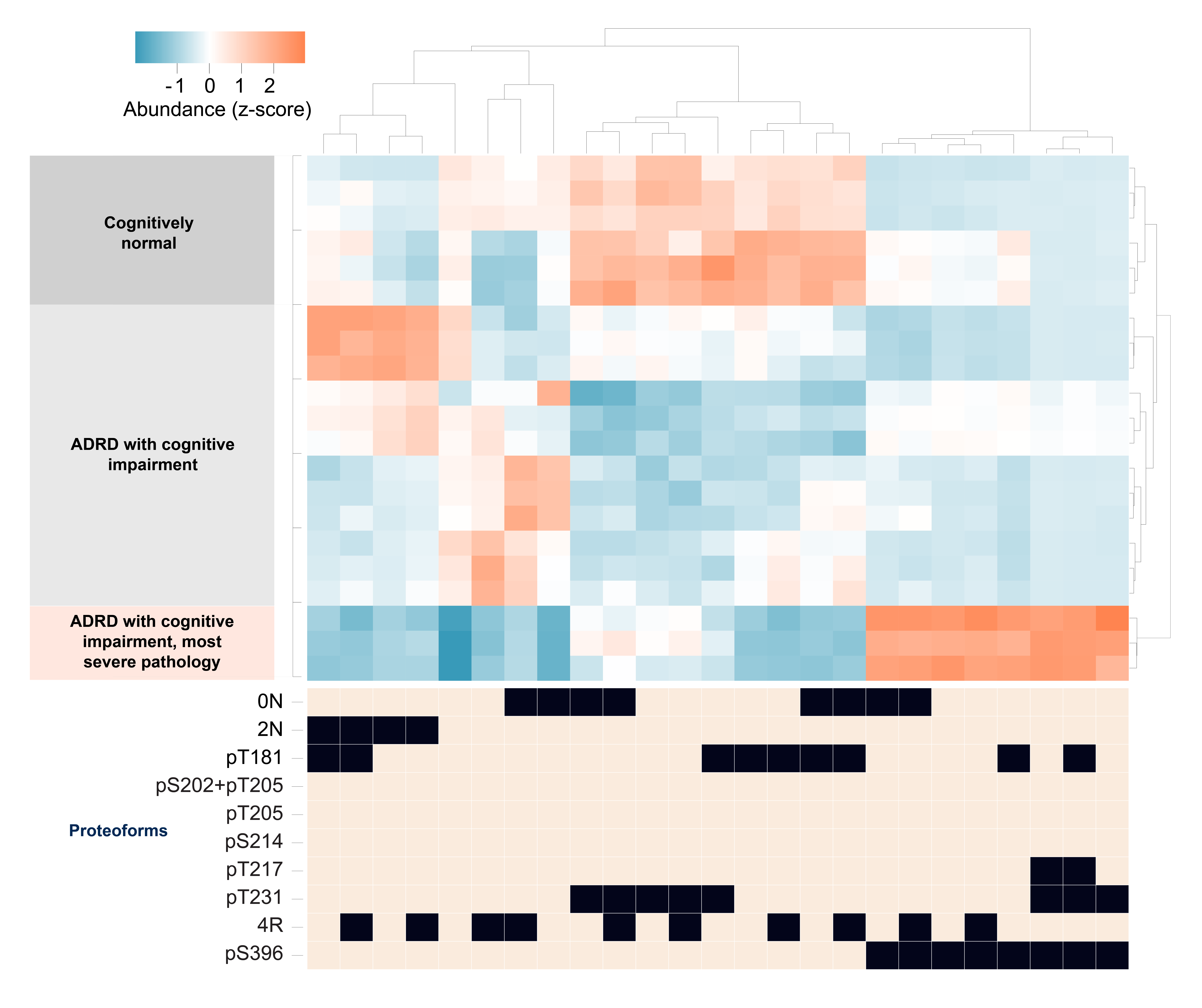
It is incredibly exciting that our platform reveals new facets of tau biology, but these findings rest on decades of prior knowledge. There are likely many similar cases where we can measure well-studied proteins’ proteoforms and quickly identify critical and novel aspects of their biology (and we’d like to work with you to study those proteoforms!). Yet, if we want to understand all diseases and all biological functions, we’ll need more data about more proteins and their proteoforms, we’ll need it at scale, we’ll need it over time, and we’ll need it under a wide variety of conditions.
What we’ll end up with are heat maps that get very complicated very quickly – hierarchical clustering and the human eye will not be enough to decipher these heat maps and associate their nuances with biological functions and diseases. Picture the tau heat map above, but millions, potentially billions of times more complex. This is not something humans can efficiently analyze. AI (and more specifically machine learning) can.
Thus, we believe it is machine learning algorithms with human supervision that will parse the important differences between proteomes needed to drive the future of biology.
We need better proteomic data to train models of biology
To model biology, machine learning algorithms will need better proteomic data than what’s available today. Current data comes from a wide variety of technologies that often detect many proteins, but don’t deliver the same qualitative or quantitative results. For instance, recent comparison studies show that popular plasma proteomics technologies measure minimally overlapping sets of proteins and produce protein abundance measurements that do not correlate well with gold-standard MS measurements using internal standards (Krisher et al., 2025).
We cannot compare studies and use them to train models of biology when they don’t measure the same proteins and worse still, don’t produce results that align with the only ground truth we’ve got (MS with internal standards). On top of that, even within a single technology, protein abundance measurements lack precision. This imprecision increases noise in what is already a very noisy part of biology and will likely make it hard for machine learning algorithms to detect small but important changes in protein networks.
That’s not to say current technologies aren’t useful – studies that use the same technologies are often comparable. These identify correlations between “proteins” or “protein groups” as defined by small sets of epitopes or peptides respectively, and biological functions whose nuances can be explored and verified through follow-up studies. But to decrease the amount of follow-up required, we need to train models that accurately represent protein molecules and predict outcomes.
Ultimately, such training will require that we know what these “protein” measurements represent at the molecular level. We need to define “proteins” by more than small sets of peptides or epitopes that might be derived from heterogenous and functionally distinct molecular entities. We can’t accept a flattening of protein heterogeneity though these measurements because machine learning algorithms may need this heterogeneity to accurately model biology. Thus, we need technologies that provide richer, more discriminating measurements of protein molecules.
As a further benefit, the outputs of these single-molecule measurements should be easier to compare than the “protein group” or “protein” intensity measurements output by mass spectrometry and standard affinity assays. The single-molecule protein and proteoform counts designed to be output by next-generation technologies like the Nautilus Platform (which is powered by Iterative Mapping) are direct representations of biology. They provide standardized, digitized units of well-defined molecules that can be measured in the same way across biological systems.
In contrast, “protein group” quantifications provided by mass spectrometry are derived from the identification and quantification of peptides that come from many different protein molecules and often different protein species. Similarly, “protein” intensity measurements provided by standard affinity assays are derived from reagent binding to whatever molecules contain the reagent’s target epitope (which may itself be nonspecific). Because of this, both mass spectrometry and standard affinity assays provide amalgamated abundance measurements coming from many different protein species that cannot be differentiated. These “bulk” measurements generate blurry and imprecise representations of proteomic landscapes that may lack the nuance and clarity needed to compare them and train new models of biology.
In the next blog post in this series, we’ll dive further into the ways data generated on the Nautilus Proteome Analysis Platform is designed to well-suited for AI workflows. Subscribe to the Nautilus Blog so you don’t miss it!
MORE ARTICLES
