
Functional differences in the KRAS proteoform landscape

Tyler Ford
September 4, 2025
If you’ve read our recent preprint, you know there may be vast proteoform landscapes hiding behind aggregated “protein” abundance measurements. Indeed, we hope that researchers can leverage Iterative Mapping of proteoforms on the Nautilus Platform to chart these landscapes and associate proteoform profiles with biological processes, disease states, and treatment outcomes. In this way, we hope proteoforms will become targets for next-generation precision medicines and biomarkers.
However, you may wonder what evidence we have that proteoforms behave differently at the molecular level. In our preprint, we saw preliminary signs that unique proteoform profiles are associated with different sample types and levels of Alzheimer’s disease severity but did not characterize the molecular functions of the proteoforms identified. Thankfully, top-down mass spectrometry researchers have provided this level of characterization for a small subset of proteoforms – their work provides strong evidence for the importance of proteoforms and lays the groundwork for future studies with more accessible and higher-throughput methods like our own Iterative Mapping of proteoforms.
In this blog post we cover the work of Adams et al., 2023 as an example of research tying a novel proteoform to a change in molecular function. This work comes from the lab of proteoform expert Neil Kelleher. His lab developed a method coupling immunoprecipitation and top-down mass spectrometry (IP-TDMS) to study KRAS proteoforms in 2018 (Ntai et al., 2018), and Adams et al. 2023 optimized this protocol characterizing even more KRAS proteoforms with an improved limit of detection. After identifying a novel truncated KRAS proteoform group prevalent in tumor samples, they used various cell and molecular biology techniques to determine how its function differs from that of canonical KRAS.
KRAS – a signaling protein with many proteoforms
KRAS is a well-studied protein that is often mutated in cancer. It is membrane-associated and drives downstream kinase signaling. Many of its post translational modifications (PTMs) have been studied and are known to alter signaling. Yet, only recently have researchers begun to identify the combinations of modifications found on KRAS proteoforms and associate them with biological outcomes. This work is critical because studying individual PTMs doesn’t capture biological reality – in live cells, PTMs will come in combinations that can only be studied and understood at the proteoform level.
A top-down proteomics pipeline for identifying KRAS proteoforms
In top-down proteomics, intact proteins are analyzed by liquid chromatography mass spectrometry (LC-MS). For their KRAS workflow, Adams et al., 2023 did the following:
- Immunoprecipitated KRAS from cell lines and primary tumor samples – immunoprecipitation simplified downstream analyses by largely limiting the analytes that entered the LC-MS to KRAS variants.
- Identified putative KRAS proteoforms by analyzing the samples with LC-MS in “discovery” mode – Here only the “MS1” scans showing the mass over charge (M/Z) peaks from intact proteoforms were identified. The result of this analysis was a series of mass spectra representing the intact proteoforms that came off the LC-column at different times. Each unique, intact proteoform ideally has its own M/Z peak in these kinds of MS1 spectra, but there may be proteoforms that have overlapping peaks that cannot be distinguished.
- Conducted targeted analysis of the proteoforms identified in discovery mode – here proteoforms defined by their specific retention times and unique M/Z peaks in the MS1 spectra were isolated and fragmented. The resulting “MS2” spectra displayed the abundances of proteoform fragments with various M/Z and were used to define the molecular make-up of each unique MS1 proteoform. Often, phosphorylations, truncations, and other posttranslational modifications making up a given proteoform can be deciphered from MS2 scans, but sometimes the positions of specific PTMs remain ambiguous even after fragmentation resulting in incompletely characterized proteoforms.
Following this workflow, Adams et al., 2023 identified 39 completely characterized and additional incompletely characterized proteoforms. Some of the modifications found on these proteoforms included:
- Acetylation
- Carboxymethylation
- Methylation
- Geranylgeranylation
- Farnesylation
- Phosphorylation
- Oxidation
- Nitrosylation
- Truncation
In addition, the authors were able to measure the abundance of proteoforms derived from KRAS mutant alleles compared to wild type alleles as well as the abundance of KRAS proteoforms derived from different KRAS isoforms. In many cases, differences in the abundances of these various proteoform groups could not be explained by corresponding differences in mRNA levels as determined by RNA-seq. This speaks to the inadequacy of RNA levels to predict protein levels.
Function of a novel truncated KRAS proteoform
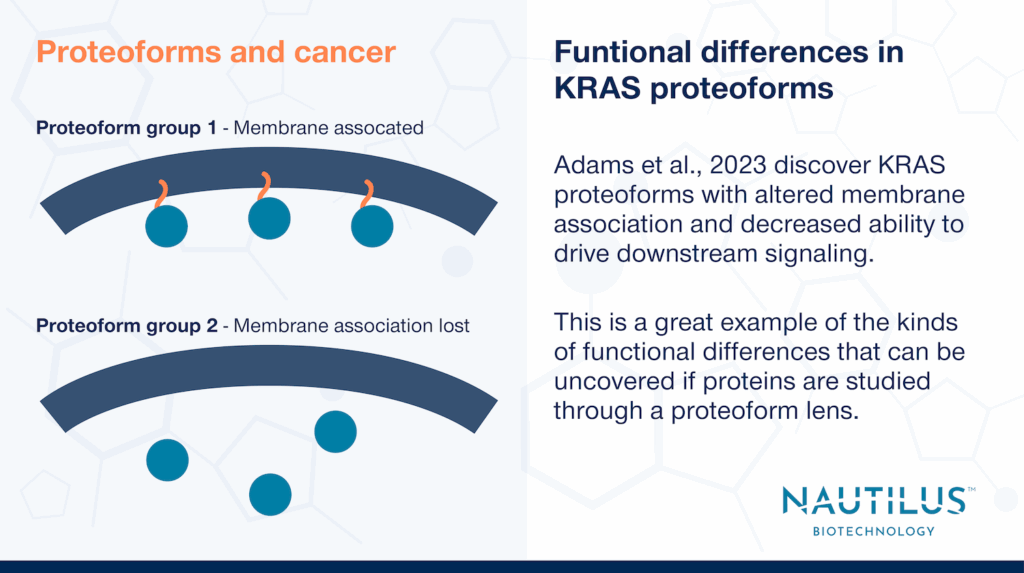
In most of the tumor samples and just one of the cell lines examined, Adams et al., 2023 discovered a group of truncated proteoforms with C185 removed. KRAS is normally farnesylated at this site and carboxymethylated at its c-terminus but the removal of C185 precludes these modifications. When found, these truncated proteoforms were often in high abundance compared to other KRAS proteoforms.
Given that farnesylation, carboxymethylation, and other modifications are important for KRAS membrane association, Adams et al., hypothesized the truncated proteoforms may not localize to the membrane and may be defective in driving signaling. They tested this by fusing a recombinant KRAS with a premature stop codon at C185 (thereby generating the truncation) to GFP and assessing its localization as well as its ability to drive ERK phosphorylation (downstream signaling). The recombinant KRAS neither localized to the membrane nor drove phosphorylation of ERK whereas wildtype fusions did. The authors posited that, given their defects and prominence in colorectal cancer tumors, these truncated proteoforms may arise as cells adapt to and counter-act proliferative, oncogenic signaling through the KRAS pathway. They further suggested the truncated proteoforms may act as dominant negative inhibitors, but more work must be done to test this hypothesis.
Future work on KRAS proteoforms
With their small number of tumor samples, these authors did not observe significant associations between any proteoforms and colorectal cancer severity. Nonetheless, their results do show that there are many KRAS proteoforms, and at least one group of them likely has altered molecular activity. Future studies may parse the roles of the additional proteoforms prioritized by their association with physiological states.
Overall, these results show the KRAS proteoform landscape is complex, and different KRAS proteoforms can adopt different functions. The next step is to associate various KRAS proteoform profiles with specific phenotypes. Such work will require more comprehensive, quantitative, and high-throughput analysis, but will hopefully reveal how mixtures of proteoforms drive phenotypic outcomes.
While KRAS is difficult to drug, researchers may be able to use their knowledge of its proteoform landscape to promote the creation of KRAS proteoforms associated with healthy outcomes or identify proteoform profiles indicative of therapeutic response. In this way knowledge of KRAS proteoforms could drive drug development and treatment decisions.
How can Iterative Mapping of proteoforms contribute to our knowledge of KRAS?
Top-down mass spectrometry does a great job of discovering new proteoforms and providing course views of the proteoform landscape. Now that many of the combinations of modifications found on KRAS proteoforms have been identified with mass spectrometry, researchers can prioritize their robust, quantitative analysis across diverse cell and tissue samples using Iterative Mapping of proteoforms.
Iterative Mapping resolves and quantifies proteoforms at the single-molecule level and does so while analyzing millions to billions of protein molecules at once. As we’ve demonstrated for tau, researchers can use Iterative Mapping to confidently quantify proteoforms over a wide dynamic range with high sensitivity. In this way, Iterative Mapping enables researchers to accurately interrogate the relative differences in these proteoforms across sample types and produce fine-grained proteoform profiles.
Our tau data suggests these high fidelity proteoform profiles provide uniquely informative views of the biology underlying diverse phenotypes. Top-down mass spec provides the outline of the landscape and Iterative Mapping can fill in all the important details needed to navigate it.
MORE ARTICLES
