
Advantages of Iterative Mapping – Comprehensive proteomic analysis

Tyler Ford
January 29, 2026
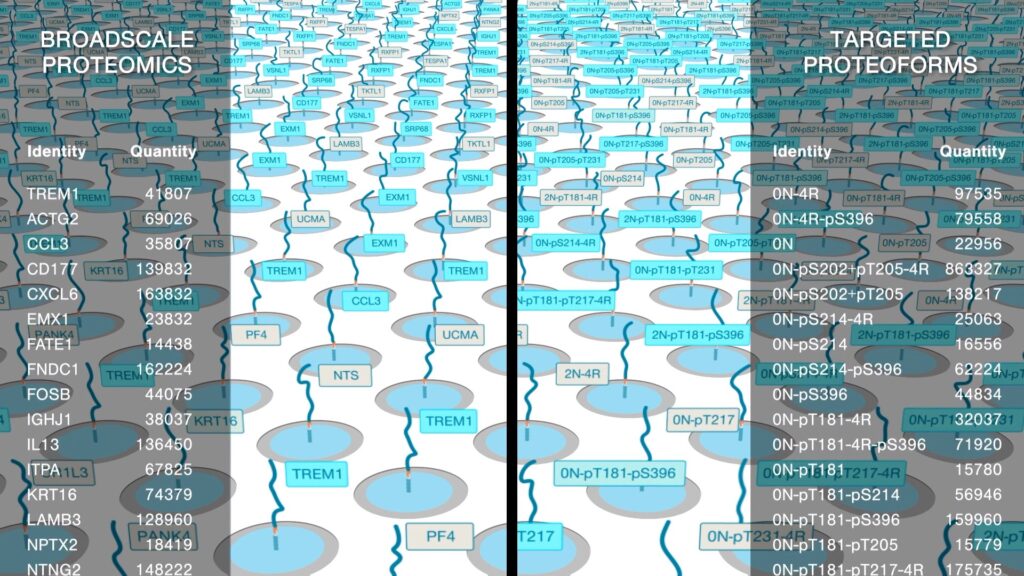
A platform with comprehensive proteomic analysis capabilities can quantify dynamic and nuanced abundance differences in proteins across the full proteome while also quantifying proteoforms – the many variants of individual protein molecules. Many proteomic analysis methods are touted for their ability to achieve broad proteome coverage, but these methods often can only detect, not quantify proteins, and can rarely detect or quantify proteoforms.
Iterative Mapping is designed to achieve comprehensive proteomic analysis with broad coverage and exceptional detail through the following abilities:
Iterative Mapping is designed to identify all proteins in proteome at the single molecule level regardless of…
Protein composition
While other methods may be challenged by the incredible compositional and chemical diversity of the proteome, Iterative Mapping leverages this diversity for protein identification. The repeated single-molecule probing that underlies Iterative Mapping detects diverse features found on each protein molecule. Machine-learning powered algorithms then assess the combinations of detected features to identify proteins. Molecules identified as the same protein or proteoform are simply counted for quantification.
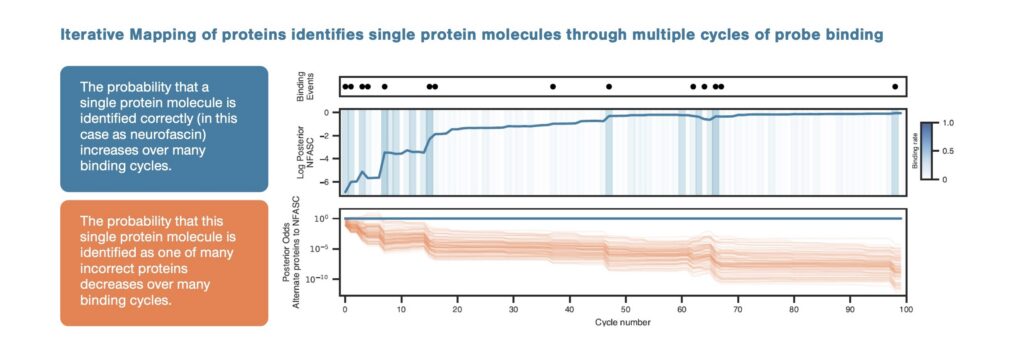
Many cycles of probe binding reveal features found on each individual protein molecule and increase the probability of correct single-molecule identification while the probability of incorrect identification decreases dramatically. Source: Data from US HUPO 2026.
Protein abundance
Iterative Mapping is conducted on massive arrays designed to capture every protein molecule in a sample. These arrays have 10-billion single-molecule protein landing pads that should not be swamped out by abundant proteins in most samples. In addition, our protein preparation and deposition techniques are designed to ensure each landing pad contains exactly one protein – with no space wasted, we expect to maximize coverage and achieve a wide dynamic range. Finally, Iterative Mapping identifies proteins through independent, single-molecule probe-protein binding interactions. The presence of other protein molecules on the array, whether they be the same or different protein molecules, is expected to have little impact on these single-molecule interactions.

Source: Data from US HUPO 2026.
Iterative Mapping is designed to quantify minute differences between proteins in terms of…
The molecular composition of individual protein molecules
The repeated single-molecule probing underlying Iterative Mapping enables it to identify epitope-defined features found on each protein molecule. In broadscale proteomic analysis, this means the method can output single-molecule binding maps indicating where probes targeting ~3-amino acid sequences likely did or did not bind every molecule. In targeted proteoform studies, this means Iterative Mapping can identify combinations of proteoform-defining features such as splice sites, post translational modifications, or any other alterations.
The abundance of molecularly defined proteins and proteoforms
Iterative Mapping is designed to identify proteins and proteoforms through molecular features probed at the single-molecule level. Individual molecules identified as the same protein or proteoform are counted for quantification. Thus, Iterative Mapping is designed to quantify single-molecule differences in protein abundance and should be able to detect minute differences in the proteomic landscape that are hidden from other methods.
Incorporate Iterative Mapping into your workflows
No other proteomics platforms available today are designed from the ground up for comprehensive proteomic analysis with broad coverage and exceptional detail. If you’d like to learn how you can incorporate the Nautilus VoaygerTM Platform and Iterative Mapping into your workflows, join our Early Access Program.
MORE ARTICLES
